Single Nucleus Pituitary Atlas
Notice: This web app is under active development at the moment. You may encounter disruption of usage and incomplete features.
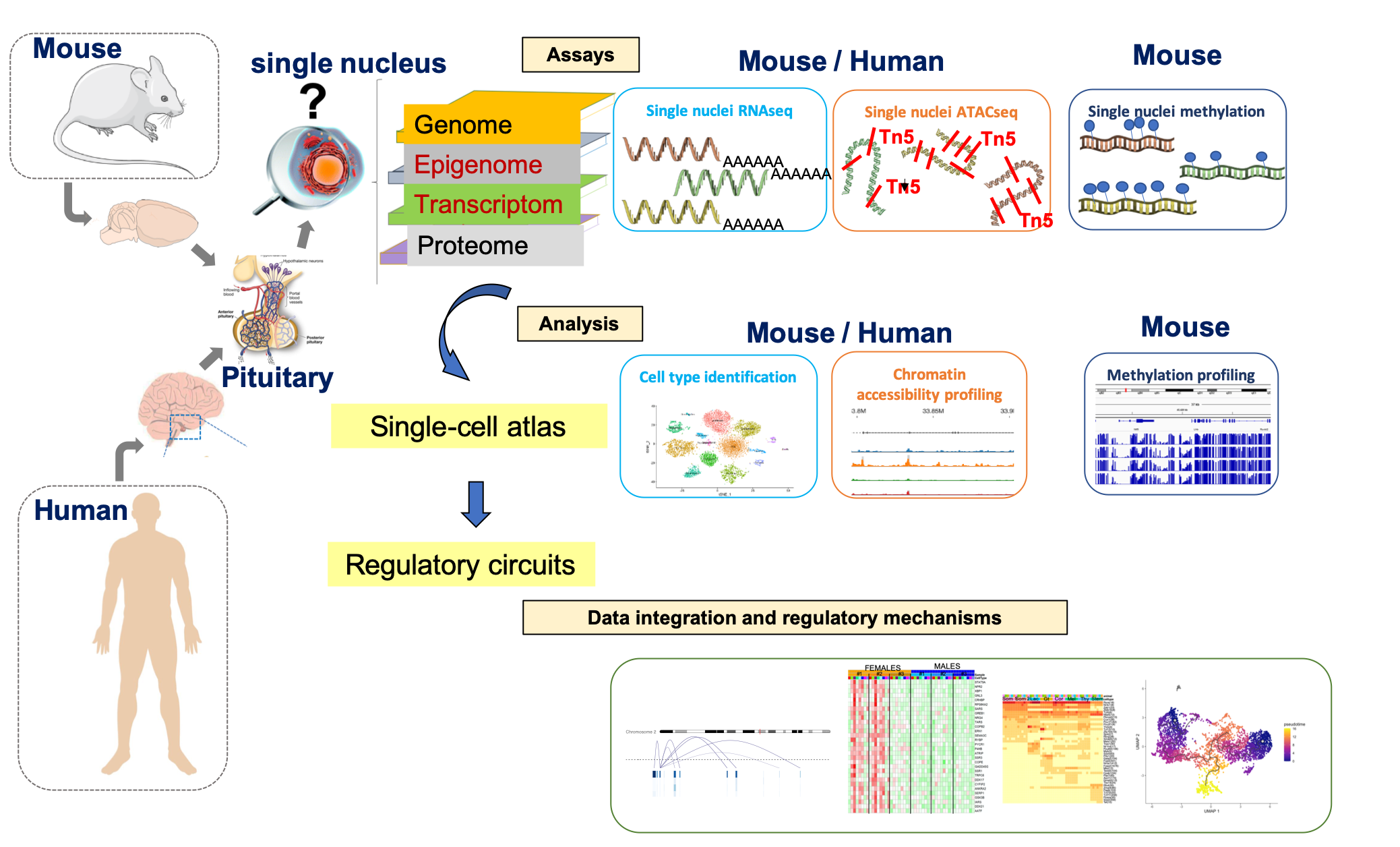
Data Browser
Browse the expression, chromatin accessibility and methylation profile of your gene of interest, either at single cell level or cell type level.
Inferred cis-Regulatory Regions
Browse the potential cis-regulatory regions of your gene of interest. The cis-regulatory regions were inferred from co-accessibility between distal elements and promoters, calculated using the CICERO package.
Regulons
Browse the regulons (TF and its potential target genes) reconstructed from the snRNAseq data. The regulons were reconstructed using the SCENIC pipeline.
Data Browser
Coembed
shows the UMAP overlay of all the cells from all selected samples and assays. Note that currently the single nucleus methylation data is only available for mouse and is pooled from 30 male animals.
Gene browser
shows the RNA expression, chromatin accessibility (ATAC) and methylation profiles of your choice of gene. It shows the gene at single-cell resolution in the same UMAP plot. It also shows at cell-type resolution in the format of violin plot for gene expression and bigWig tracks for chromatin accessibility and methylation profiles. Note that currently the methylation profile is only available for mouse.
Coembed
Single Cell Gene Browser
RNA
ATAC
Methylation
Cell Type Gene Expression
Cell Type ATAC and Methylation
Inferred cis-Regulatory Regions
Browse the potential cis-regulatory regions of your gene of interest. The cis-regulatory regions were inferred from co-accessibility between distal elements and promoters, calculated using the CICERO package.
Gene-centered Putative Regulatary Regions
Regulons analysis
A regulon is defined as a set of genes including one transcription factor (TF) and all its potential target genes. The regulons were reconstructed using the SCENIC pipeline. You can select a regulon (represented by the name of the TF) to explore its enrichment in individual cells or cell types.
Top regulons in each cell type
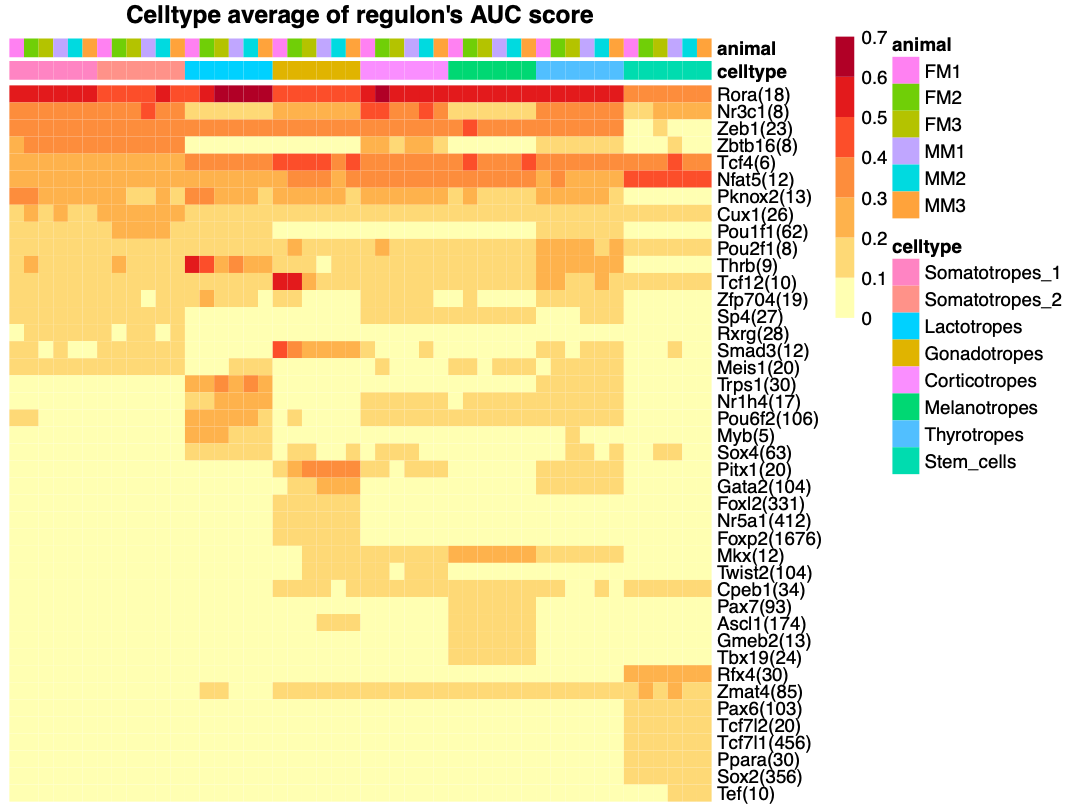
Regulon activity at single-cell resolution
Regulon info
Regulon activity at cell-type resolution
Introduction
Cite
Single nucleus multi-omics regulatory atlas of the murine pituitary
Frederique Ruf-Zamojski, Zidong Zhang, Michel Zamojski, Gregory R. Smith, Natalia Mendelev, Hanqing Liu, German Nudelman, Mika Moriwaki, Hanna Pincas, Rosa Gomez Castanon, Venugopalan D. Nair, Nitish Seenarine, Mary Anne S. Amper, Xiang Zhou, Luisina Ongaro, Chirine Toufaily, Gauthier Schang, Joseph R. Nery, Anna Bartlett, Andrew Aldridge, Nimisha Jain, Gwen V. Childs, Olga G. Troyanskaya, Joseph R. Ecker, Judith L. Turgeon, Corrine K. Welt, Daniel J. Bernard, Stuart C. Sealfon
bioRxiv 2020.06.06.138024; doi:
https://doi.org/10.1101/2020.06.06.138024
Single nucleus pituitary transcriptomic and epigenetic landscape reveals human stem cell heterogeneity with diverse regulatory mechanisms
Zidong Zhang, Michel Zamojski, Gregory R. Smith, Thea L. Willis, Val Yianni, Natalia Mendelev, Hanna Pincas, Nitish Seenarine, Mary Anne S. Amper, Mital Vasoya, Venugopalan D. Nair, Judith L. Turgeon, Daniel J. Bernard, Olga G. Troyanskaya, Cynthia L. Andoniadou, Stuart C. Sealfon, Frederique Ruf-Zamojski
bioRxiv 2021.06.18.449034; doi:
https://doi.org/10.1101/2021.06.18.449034
Funding
This work was supported by funding from the National Institute of Health (NIH) Grant DK46943 (SCS), the Canadian Institutes of Health Research (CIHR) Project Grants PJT-162343 (DJB) and PJT-169184 (DJB), NIH award R01HD065029 from the Eunice Kennedy Shriver National Institute Of Child Health & Human Development (CKW), NIH NICHD R01HD093461 (GVC), NIH R01HD087057 (GVC), and NIH NIDDK 1R01DK113776-01 (GVC).